Friday, 18 May 2007
3rd Floor Hall (Pfahler Hall)
473
Density functional theory study of the two electron oxidation of 3,4-dimercaptobenzene-1,2-diol
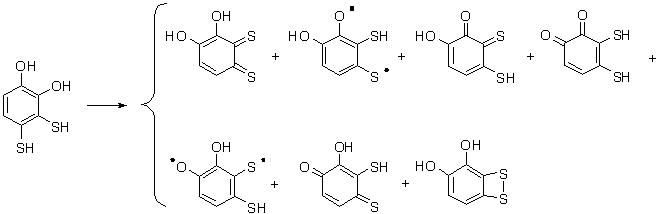
Oxidation of 3,4-dimercaptobenzene-1,2-diol is examined by density functional theory methods to predict the outcome of a two electron oxidation. Various structures could be anticipated (see the Scheme below). Our study focused on the likelihood for the production of the quinone, thioquinone, and dithioquinone compounds that might be expected to arise. Suitable agreement is found between computed isodesmic reactions for DH0f (gas) and the experimentally derived DH0f (gas) values for the parent o-, m-, and p-benzoquinones. Our DFT computations initially suggest benzo[c][1,2]dithiete-3,4-diol to be the energetically preferred oxidation product. Formation of p-thioquinone is slightly less favored, while o-quinone, o-thioquinone o-dithioquinone and the m-thioquinone diradicals are predicted to be considerably less stable.
Back to Poster Session V
Back to The Middle Atlantic Regional Meeting (May 16 - 18, 2007)